 复旦主页
复旦主页 实验室安全
实验室安全 复旦邮箱
复旦邮箱 办事大厅
办事大厅



近期,复旦大学信息科学与工程学院张浩副教授团队提出了创新的通用原子嵌入方法(ct-UAE),该方法基于自主研发的 CrystalTransformer 模型,在晶体材料物性预测领域取得了显著进展。研究成果不仅有效提高了形成能、带隙和力等重要物理属性的预测精度,并且通过在大规模数据库上进行多任务学习,ct-UAE展现了强大的知识迁移能力,能够保持在数据稀缺任务中的良好泛化性能,解决了传统方法在小数据集上的应用瓶颈。该研究成果于2025年1月31日在线发表于《Nature Communications》。
随着机器学习在材料科学中的广泛应用,材料属性的预测已经成为推动科技进步的重要方向。传统的预测方法大多依赖于人工设计的输入特征和简单的模型,但这些方法在面对复杂的材料系统时常常面临数据稀缺和预测精度有限的问题。近年来,深度学习尤其是图神经网络(GNN)在材料科学中的应用取得了显著进展,然而,如何有效表征原子间的复杂相互作用并提高预测精度,仍然是一个亟待解决的挑战。
在这种背景下,原子嵌入方法作为深度学习模型中的关键步骤,起到了连接材料的化学信息和物性特征的桥梁作用。然而,现有的原子嵌入方法大多依赖于预定义的特征或人工设计的嵌入方式,难以适应不同材料体系和任务的需求。对于这一缺陷,基于深度学习的新型嵌入方法能够通过自动学习原子及其相互作用的高维特征,提升材料属性预测的精度和泛化能力。
近几年,基于 Transformer 架构的模型在自然语言处理领域取得了革命性进展,逐渐引入到材料科学中。通过对原子信息进行嵌入并结合图神经网络进行学习,研究者们取得了突破性进展。然而,现有方法在面对数据稀缺任务时,仍然存在挑战。如何在大规模数据库上进行有效训练,并确保方法在数据不足的情况下依然能够保持良好的预测精度,是当前材料科学中悬而未决的重要问题。
为了应对这些挑战,我们提出了一种基于自主研发的CrystalTransformer 模型的通用原子嵌入方法(ct-UAE)。该方法通过自动学习原子嵌入,不依赖于预定义的原子特性,能够适应多种材料的属性预测任务。我们还通过多任务学习,在大数据库中训练获得的知识能够有效迁移至钙钛矿等数据稀缺任务,大大提高了模型的泛化能力。我们的研究为解决数据稀缺问题和提升预测精度提供了新的思路,具有广泛的应用前景。
该工作提出了一种基于 CrystalTransformer 模型的通用原子嵌入方法(ct-UAE),通过针对性设计的前端模型CrystalTransformer在大规模数据库中进行训练,隐式地获得表现良好且具有优良可迁移性的原子表征,即原子嵌入向量,再迁移到各种后端模型上对不同的目标物性进行预测,取得良好的结果。
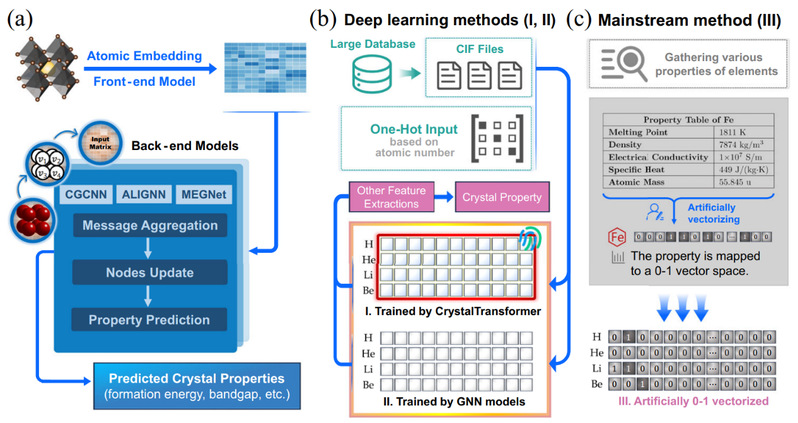
图1 通用原子嵌入方法的工作流程图。基于CrystalTransformer模型训练的通用原子嵌入与利用其他图神经网络训练的结果,以及传统的人工设计的嵌入方式是三种不同的嵌入方式。
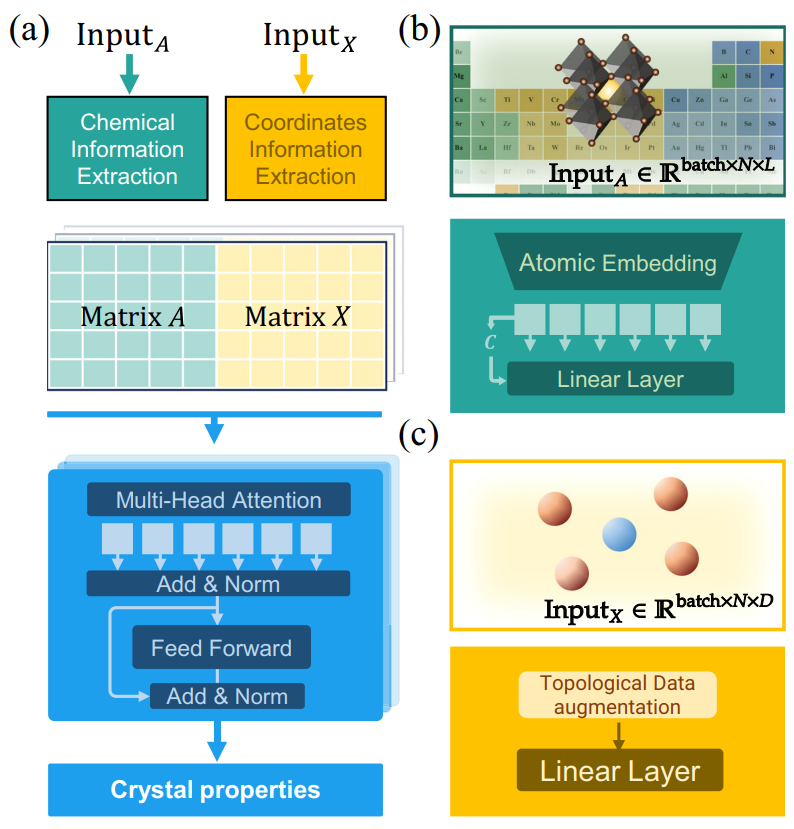
图 2 针对性设计的CrystalTransformer的模型架构。CrystalTransformer避免使用了图神经网络的基本架构,减少固有的归纳偏置对晶体拓扑结构的关注,利用Transformer的自注意力机制充分提取晶体中隐含的原子化学信息。
图1给出了基于CrystalTransformer模型训练的通用原子嵌入与利用其他图神经网络训练的结果,以及传统的人工设计的嵌入方式是三种不同的嵌入方式的对比,针对性设计的CrystalTransformer由图2给出。作为前端模型,CrystalTransformer的作用在于从大规模晶体数据中充分提取出与原子化学信息相关的那一部分并融入原子嵌入向量中,不使用图神经网络作为基本架构,减少了归纳偏置对晶体拓扑信息的关注,并利用Transformer架构充分提取原子本身的化学信息。
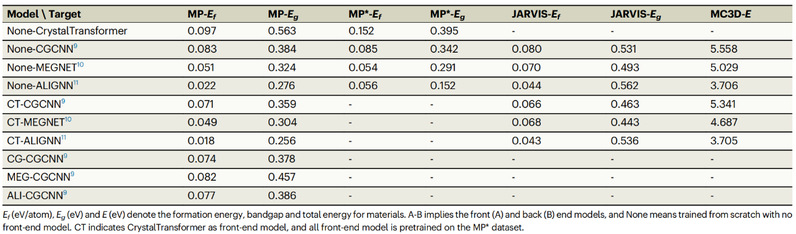
表1 不同前端-后端模型组合在不同任务中的预测结果对比。使用CrystalTransformer作为前端模型的综合预测结果最佳。
表1展示了不同前端-后端模型组合的预测结果对比,在MP、MP*、JARVIS以及MC3D数据集中的形成能、带隙以及总能等物性上进行了测试,使用CrystalTransformer作为前端模型的预测精度最高,误差最低。
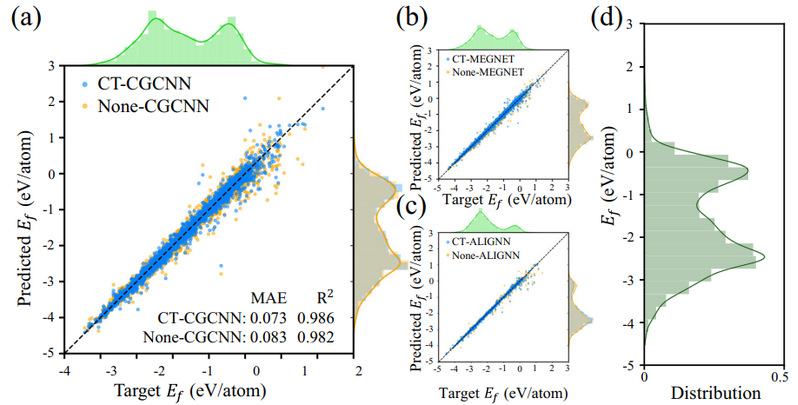
图3 在CGCNN、MEGNET和ALIGNN三种图神经网络中使用通用原子嵌入与否的结果。使用CrystalTransformer作为前端模型,即使用了通用原子嵌入的结果有明显提升。
图3展示了在CGCNN、MEGNET和ALIGNN三种流行的图神经网络中,使用了通用原子嵌入的模型在形成能任务中取得了预测精度的明显提升,在CGCNN模型中,使用通用原子嵌入的MAE从0.083下降为0.073。此外,对于力、应力等矢量物性预测任务,将通用原子嵌入迁移到CHGNet、M3GNet和MACE中,预测效果也有所提升,CHGNet预测力任务的损失从0.284下降为0.242,提升了约14.8%的预测精度。
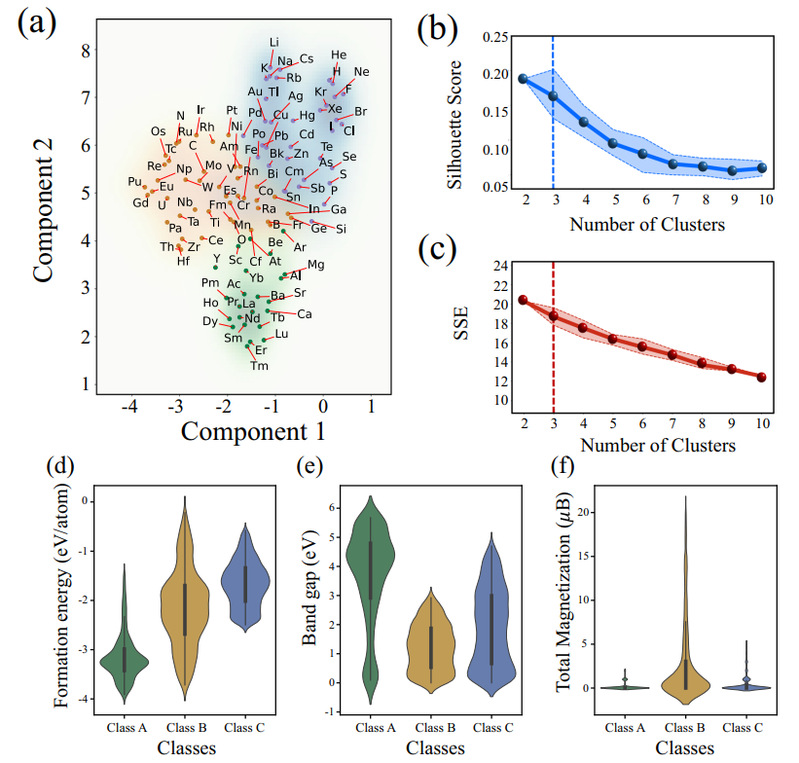
图4 将通用原子嵌入进行聚类的示意图。聚类结果表现出物理上的可解释性。
为了验证通过训练CrystalTransformer模型提取出的通用原子嵌入充分反映出原子的化学本质,从而在物理化学相关任务中是“通用的”,即良好的可迁移性,我们使用UMAP算法和K-means算法分别对通用原子嵌入进行了降维与聚类,将其聚类为3类的结果由图4给出。这一聚类结果是模型对于所选定的形成能、带隙、总能和总磁化强度几种物性综合学习的结果,不仅仅反映了原子本身在元素周期表中的排列所揭示的电负性、半径等规律,更融合了原子在晶体中的所发挥的不同作用的相关信息。我们筛选MP中所有的仅包含上述聚类的3类元素与氧化合的所有氧化物(及氧单质),分析了其在形成能、带隙和总磁化强度上的分布规律,发现其呈现出显著的差异。A类元素所形成的氧化物的形成能较低,集中在−2.5 eV/atom至−4.0 eV/atom之间,表明具有较高的化学稳定性,带隙介于3 eV至6 eV之间,主要为宽带隙半导体。B类元素的氧化物形成能范围较广,从接近0 eV/atom到−4 eV/atom,带隙分布在0.5 eV至2.5 eV之间,表现出窄带隙半导体特性,具有较低的化学稳定性。C类元素所形成的氧化物的形成能集中在−1.0 eV/atom至−2.5 eV/atom之间,带隙则介于1 eV到4 eV之间,表现出较好的化学稳定性。此外,大多数氧化物的磁化强度接近0 ![]() B,显示出顺磁性或抗磁性,B类元素中的部分氧化物表现出较强的铁磁性。
B,显示出顺磁性或抗磁性,B类元素中的部分氧化物表现出较强的铁磁性。
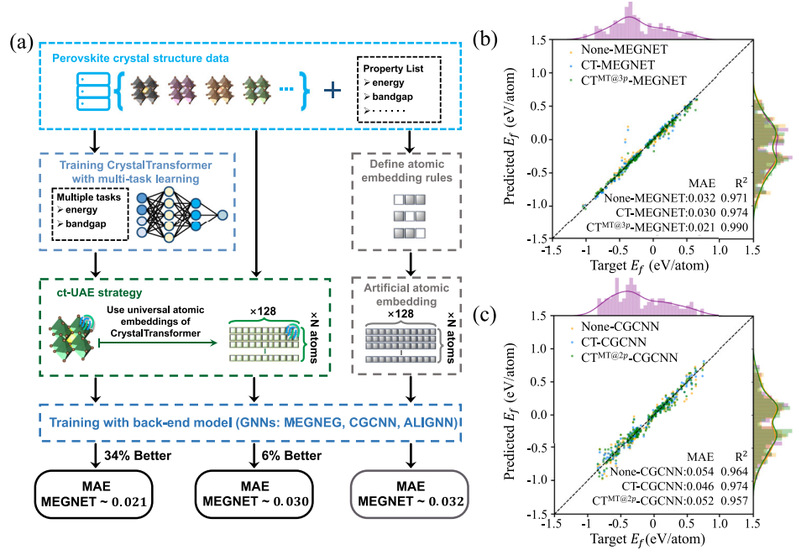
图5 将通用原子嵌入用于钙钛矿物性预测任务中的流程及预测效果。使用多任务学习的通用原子嵌入表现出了最高的预测精度
验证了具有良好可迁移性和物理可解释性的通用原子嵌入应当能够在数据稀缺的任务中保持模型的泛化性能和预测精度,而不至于由于数据稀缺带来的欠拟合等导致较高的预测误差。钙钛矿的物性预测是数据稀缺的例子,图5展示了在钙钛矿晶体的形成能上迁移使用多任务学习的通用原子嵌入的效果,在MEGNET上,预测的MAE从0.032下降到0.021,预测精度提升了34%,说明通用原子嵌入从大规模数据集迁移的通用知识在很大程度上帮助了钙钛矿材料物性等数据稀缺任务的预测过程,从而提升了模型的泛化性能。
该研究提出的 ct-UAE 方法通过训练自主研发的 CrystalTransformer 模型,提取出具有高精度、优良可迁移性和物理可解释性的通用原子嵌入,提升了晶体材料属性预测的准确性,并有效解决了传统方法在数据稀缺任务中的挑战。通过多任务学习,ct-UAE在大规模数据库上训练获得的知识能够有效迁移至数据稀缺任务,展现出良好的泛化能力和跨任务转移性能。该方法在形成能、带隙和力等多个材料属性预测中均取得了显著提升,且在实际应用中具有较强的扩展性和适用性。本项工作为提升模型在数据稀缺任务中的泛化性能提供了创新的思考角度,且十分基础。
复旦大学本科生金罗智杰与本科生杜子健是论文共同第一作者,复旦大学张浩副教授、岑剡博士(高级工程师)为共同通讯作者。复旦大学梅永丰教授与山东建筑大学许园风副教授为这项工作做出了重要贡献。
以上工作获得国家重点研发计划项目、上海市自然科学基金项目与复旦大学本科生学术研究资助计划(FDUROP)支持。
文章链接:https://www.nature.com/articles/s41467-025-56481-x